
Validation de méthodes analytiques
Dernière modification : 05.11.2024
Naviguer dans ce dossier
- Pour rendre plus aisée la navigation dans ce dossier un peu long, vous pouvez accéder directement à chaque grande partie :
- Les enjeux
- L'incertitude analytique
- Les préalables indispensables
- Décrire les méthodes et modes opératoires
- Les références méthodologiques
- Les références en biologie moléculaire
- Le processus de validation
- Les différents types de méthodes analytiques – et les dossiers de validation associés
- Un petit peu de statistiques
- La confirmation en routine
Les enjeux de la validation de méthodes analytiques
 Pour un laboratoire, il est important d'avoir confiance dans la validité des résultats analytiques qui sont rendus aux clients. C'est vrai dans le sens inverse : le client a besoin d'avoir confiance dans les résultats qu'il a sous-traités. La reconnaissance de la compétence des laboratoires est notamment attestée par leur accréditation par le Cofrac selon les normes ISO 17025 ou 15189.
Pour un laboratoire, il est important d'avoir confiance dans la validité des résultats analytiques qui sont rendus aux clients. C'est vrai dans le sens inverse : le client a besoin d'avoir confiance dans les résultats qu'il a sous-traités. La reconnaissance de la compétence des laboratoires est notamment attestée par leur accréditation par le Cofrac selon les normes ISO 17025 ou 15189.- À ce propos, depuis le 13 janvier 2010, les Laboratoires de Biologie Médicale français doivent être accrédités selon la Norme ISO 15189, qui prévoit de manière explicite la validation des méthodes analytiques. Voir la page que je consacre à ce sujet des laboratoires de biologie médicale.
- Le client, quant à lui, fait confiance au laboratoire ... jusqu'à ce que cette confiance soit reprise - ce qui arrive dès la première fois où il peut remettre en cause la compétence de son sous-traitant.
- Mais parfois, les résultats doivent absolument être "vrais". Notamment lorsque la sécurité des individus est en jeu. Par exemple :
- contrôle de matériaux (construction, aéronautique, automobile)
- contrôle de produits de santé (médicaments, dispositifs implantables)
- contrôle de contamination microbienne (restauration, alimentation, hygiène hospitalière)
- analyses de paramètres biologiques (lorsque l'on décide d'actions thérapeutiques au vu des résultats d'analyses)
- Dans de nombreux cas, les analyses sont coûteuses, ou alors les échantillons sont fragiles et il est difficile de les conserver en état, ou encore la quantité disponible interdit de recommencer la manipulation. Enfin, lorsqu'il s'agit de laboratoires de recherche publics, il est évident que l'excellence n'est pas négociable, ni pour le public, ni pour les organismes de tutelle.
- Tout ceci montre l'importance de pouvoir obtenir, dès le premier essai, un résultat en lequel toutes les parties intéressées pourront avoir confiance : client, chef de laboratoire, partenaire industriel, partenaire scientifique. De plus, les résultats obtenus pourront être repris plusieurs mois plus tard, sans que personne ne les remette en cause.
- En allant un peu au-delà - et c'est surtout vrai pour les activités de recherche - il est souvent primordial de ne pas pouvoir douter des résultats. Car alors on peut être tenté de rejeter comme erroné un résultat qui ne va pas dans le sens de la théorie que l'on développe, et d'accepter comme vrais ceux qui apportent les "preuves" que l'on recherche.
L'incertitude
- J'ai utilisé plus haut le mot "confiance". On en a besoin ici car, par essence, les manipulations qui sont effectuées dans les laboratoires donnent des résultats qui ne peuvent pas être absolument identiques. Chacun sait qu'il y a une incertitude, liée au caractère aléatoire des variations observées lorsque l'on répète une mesure.
- Les principaux facteurs qui contribuent à ces variations peuvent être résumés sur le diagramme d'Ishikawa ci-dessous (diagramme très simplifié) :
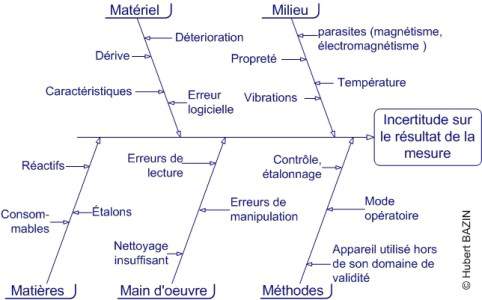
- Par deux fois au moins, des techniciens m'ont suggéré de rajouter une cause sur la branche "main d'oeuvre" : l'honnêteté des manipulateurs... Il semble donc que, au moins pour certaines équipes, ce soit une préoccupation importante, sinon majeure. Bien entendu, dans ce cas, l'objectif est de diminuer la variabilité apparente. Mais comme on ne connaît pas la valeur à atteindre, l'entreprise a des résultats très pervers : on a le sentiment d'un processus maîtrisé, mais en réalité on avance à l'aveuglette...
- Ce schéma n'est qu'une ébauche, trop généraliste pour être d'une réelle utilité pratique. Vous devez l'adapter à votre activité, votre environnement, vos analyses ou examens, etc. J'ai eu la surprise de retrouver ce diagramme, sans autre modification que la disparition de la mention de copyright, dans la procédure générale d'un très important laboratoire. Je suis au regret de devoir insister : ce n'est pas ainsi que l'on travaille !
Les préalables indispensables à la validation de méthode
- Le diagramme ci-dessus nous rappelle que la méthode elle même n'est pas la cause unique des variations dans les réultats. Si l'on veut pouvoir ne s'intéresser qu'à la méthode analytique (et c'est notre cas ici), il va falloir au préalable maîtriser les autres facteurs. Les pré-requis sont ainsi :
- Un Système de Management de la Qualité. La certification ISO n'est pas indispensable, mais l'organisation qualité, si! Il faut notamment :
- s'engager à progresser
- Définir les responsabilités
- Identifier les "clients" et leurs attentes
- Fixer des objectifs
- Se doter d'outils, dont le système documentaire et le cahier de laboratoire
- Auditer le fonctionnement
- Améliorer le système
- s'engager à progresser
- Connaître les points critiques. Avant de valider une méthode, il faut connaître les points qui sont critiques pour la qualité, c'est à dire ceux qui sont essentiels pour le client, pour le résultat, pour l'échantillon. Cela peut aller de la traçabilité à l'expression du résultat dans une unité particulière, en passant par un pré-traitement particulier de l'échantillon ou le recours à un sous-traitant spécifique pour l'interprétation du résultat brut. Ces points critiques devront absolument être maîtrisés.
- Du personnel formé, compétent. C'est bien évidemment sous-entendu dans toutes les activités de laboratoire :
- on valide les formations des personnes avant leur embauche;
- ces personnes suivent un parcours d'habilitation avant de manipuler (les résultats des manipulations de validation sont bien entendu enregistrés, voir plus haut "système qualité");
- on pourvoit, autant que de besoin, à la formation continue de l'ensemble du personnel
- on valide les formations des personnes avant leur embauche;
- Du matériel adapté : on ne pèse pas 10 µg d'un réactif sur un pèse-lettres, tout le monde le sait ! C'est vrai de tout appareil de mesure, comme de tout équipement.
On peut rappeler ici la notion de "capabilité" (notée Cp). C'est le rapport erreur maximale tolérée / (6 × écart type). On a coutue de dire que :
- Si Cp <1, l'appareil n'est pas capable (on a moins de 99,7% des valeurs à l'intérieur de la fourchette de tolérances)
- Si Cp >2, l'appareil est capable (c'est une valeur très conservatrice : on est à 99,9997 % des valeurs dans la fourchette de tolérance), on peut descendre jusqu'à 1,3, ce qui donne environ 99.98 % des valeurs dans l'intervalle de tolérance &mdask; un score plutôt honorable)
- Entre les deux, il est "juste capable", et on doit s'en méfier
On voit au passage qu'il est toujours utile d'enregistrer scrupuleusement les résultats des analyses, puisqu'ensuite on se préoccupe de l'écart-type des séries de mesures... On voit même qu'un appareil peut être "capable" au milieu de sa plage de travail, et "non-capable" quand on approche des limites de cette plage. Mais pour le savoir, il faut faire des campagnes de mesures, des répétitions, etc. C'est ce qu'on appelle la validation du matériel - préalable à la validation des méthodes qui utilisent cet appareil. - Si Cp <1, l'appareil n'est pas capable (on a moins de 99,7% des valeurs à l'intérieur de la fourchette de tolérances)
- Un suivi métrologique du matériel : on vient de voir qu'il faut utiliser du matériel adapté. Mais il faut tout autant vérifier en permanence que ce matériel est toujours adapté. En effet tout matériel vieillit, et doit ête contrôlé, entretenu, ajusté, étalonné, etc. On fait souvent contrôler les balances une fois par an par des entreprises validées (des laboratoires d'étalonnages accrédités). Mais à quelle fréquence fait-on étalonner le détecteur ECD d'un chromatographe en phase gazeuse, ou un réfractomètre, ou un banc de traction, ou un spectrophotomètre ?
Le suivi métrologique minimum consiste à dresser une liste des appareils du laboratoire, à créer une "fiche de vie" pour chacun d'eux, et à y noter toutes les opérations, toutes les questions, tous les problèmes. Ce suivi minimaliste ne permet pas d'être accrédité par le COFRAC (loin de là !), mais il permet d'avoir des données pour analyser, le cas échéant, un problème sur une manipulation. - Un environnement sous contrôle : les locaux de travail doivent être dédiés à certaines activités, avec séparation des zones de travail (pièces, hottes, postes de sécurité microbiologique si l'on travaille avec des agents pathogènes). Les pièces critiques seront climatisées, le système électrique sera régulé, les parasites électromagnétiques seront éliminés, l'eau utilisée sera de qualité maîtrisée, etc.
- Un Système de Management de la Qualité. La certification ISO n'est pas indispensable, mais l'organisation qualité, si! Il faut notamment :
Décrire les méthodes et modes opératoires
- C'est l'étape initiale du travail de validation : on écrit précisément ce que le technicien doit réaliser. A l'issue, on ne doit plus avoir le moindre doute, la moindre erreur, la moindre imprécision dans le texte. Il faut porter une attention spéciale :
- aux unités utilisées. On préfèrera toujours les unités "officielles". Par exemple, pour l'expression de la viscosité, on abandonnera définitivement le Poise ou le centiPoise, avantageusement remplacés par le milliPascal.seconde, ou mPa.s. C'est d'autant plus facile que l'on a : 1cP =1 mPa.s !.
- à l'encadrement des durées ou des températures (± x secondes , ou ± y °C)
- à utiliser la "bonne" unité : par exemple pour la centrifugation : le g (les tours par minute, ou rpm ne permettant pas de passer facilement d'une centrifugeuse à une autre, ni d'un rotor à un autre)
- aux calculs et formules utilisées
- etc.
- Si l'on souhaite procéder à une vérification de méthode analytique, c'est à dire s'assurer qu'une méthode "commerciale" donne bien les résultats attendus, présente bien les performances annoncées, alors il faudra bien entendu utiliser la méthode du fournisseur. Dans ce cas, il n'est pas toujours pertinent de ré-écrire la méthode en question.
Les références méthodologiques
- La problématique de validation de méthodes n'est pas une nouveauté; de nombreuses références ont été publiées sur le sujet :
- IUPAC Technical Report : Harmonized guidelines for single-laboratory validation of methods of analysis (Pure Appl. Chem., vol. 74, No. 5, pp.835-855, 2002.) Publié par l'IUPAC (International Union of Pure and Applied Chemistry, à la suite du Symposium sur l'harmonisation des systèmes de management de la qualité (IUPAC, AOAC, ISO) de 1999. C'est la seule référence qui traite spécifiquement de la validation par un laboratoire unique (pas de mesure de reproductibilité). Après des considérations détaillées sur la problématique, on retrouve 10 pages de recommandations, plus une bibliographie importante.
- Recommandations de l'International Counsil for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) : Validation of Analytical Procedures : text and methodology [Q2(R1)], 1994-1996. Document de 17 pages, dont seulement 7 de méthodologie. Y sont proposées plusieurs approches pour la détermination des limites de détection et de quantification (visuelle, rapport signal-bruit, écart type et pente, écart type du blanc).
- Recommandations du National Institute of Standards and Technology (NIST) : NIST Guidelines for Evaluating and Expressing Uncertainty of NIST Measurement Results (NIST technical note 1297, 1994 edition) Document interne au NIST (américain), il explique l'approche harmonisée utilisée par les multiples labos du NIST, qui ne font que de l'étalonnage; présente les méthodes de type A (approche statistique) et de type B (toute autre approche, y compris littérature, connaissances du responsable d'essai, etc.); et rappelle les lois de propagation des incertitudes.
- La série de Normes ISO 5725 Exactitude (justesse et fidélité) des résultats et méthodes de mesure, 1994 / 1996 / 2020. L'ensemble forme un corpus complet, très "statistique". C'est une référence absolue représentant près de 300 pages :
- ISO 5725 – 1 : principes généraux et définitions
- ISO 5725 – 2 (révisée en août 2020) : méthode de base pour la détermination de la répétabilité et de la reproductibilité d'une méthode de mesure normalisée
- ISO 5725 – 3 : mesures intermédiaires de la fidélité d'une méthode de mesure normalisée
- ISO 5725 – 4 (celle-ci a été révisée en septembre 2020) : méthode de base pour la détermination de la justesse d'une méthode de mesure normalisée
- ISO 5725 – 5 : mesures alternatives pour la détermination de la fidélité d'une méthode de mesure normalisée
- ISO 5725 – 6 : utilisation dans la pratique des valeurs d'exactitude. "La" référence en matière de validation statistique
- La Norme ISO 17025 : 2017 (la version précédente datait de 2005) Exigences générales concernant la compétence des laboratoires d'étalonnage et d'essais. C'est la référence pour l'accréditation des laboratoires par le COFRAC. Elle s'organise suivant 8 chapitres, et prévoit deux options pour le système de management de la qualité : soit le laboratoire est certifié ISO 9001, soit son SMQ est conforme aux exigences de l'ISO 9001... Elle exige que les méthodes soient vérifiées ou validées, sans expliquer clairement ce que l'on entend par là.
- La Norme ISO 15189 : 2022 ( elle a changé récemment) Créée à l'initiative des laboratoires d'analyses de biologie médicale, ce n'est qu'une adaptation de l'ISO 17025 : elle traite explicitement des phases pré– , per– et post– analytiques; elle attribue de nombreuses responsabilités au biologiste directeur de laboratoire; elle intègre des exigences relatives à la sécurité du personnel et aux prestations de conseil.
- Les guides du COFRAC Disponibles sur le site Internet du COFRAC: www.cofrac.fr.
- Guide technique d'accréditation de vérification / validation des méthodes en biologie médicale, SH GTA 04, révision 2 de 2023. Il distingue les méthodes quantitatives et qualitatives, mais surtout les méthodes "commerciales" des méthodes développées, ou adaptées par le laboratoire, qui doivent être validées soigneusement, alors que les premières peuvent n'être que vérifiées. Il aborde la question du contenu du dossier de validation et fournit des exemples ;
- Les contrôles de la qualité en biologie médicale, SH GTA 06 (juin 2012) S'intéresse à la manière de valider la qualité des analyses en routine. Il ne s'agit plus de valider une méthode, mais l'application d'une méthode validée par ailleurs. On y aborde le problème de la reproductibilité, des comparaisons interlaboratoires Prend des exemples en microbiologie (antibiogrammes, identifications...)
- IUPAC Technical Report : Harmonized guidelines for single-laboratory validation of methods of analysis (Pure Appl. Chem., vol. 74, No. 5, pp.835-855, 2002.) Publié par l'IUPAC (International Union of Pure and Applied Chemistry, à la suite du Symposium sur l'harmonisation des systèmes de management de la qualité (IUPAC, AOAC, ISO) de 1999. C'est la seule référence qui traite spécifiquement de la validation par un laboratoire unique (pas de mesure de reproductibilité). Après des considérations détaillées sur la problématique, on retrouve 10 pages de recommandations, plus une bibliographie importante.
Les références en biologie moléculaire
- j'accompagne les travaux de validation de méthodes de laboratoires de recherche utilisant des méthodes de biologie moléculaire. Dans ce domaine, comme dans les autres on va trouver si on les cherche de nombreuses publications sur la validation. Je ne cite celles-ci qu'à titre d'exemple.
- Les références spécifiques aux méthodes PCR Liées à la circulation des OGM (pour obtenir un agrément, la réglementation exige de pouvoir retrouver l'OGM, à moins de 1%) :
- Guide de l'ENGL (European Network of GMO Laboratories) Disponible sur le site de l'ENGL.
- plusieurs guides du Codex Alimentarius ou Directives du même Codex sur la mesure de l'incertitude, sur la validation par un seul laboratoire, et sur les méthodes d'analyses pour les "aliments dérivés de la biotechnologie".
- Norme AFNOR XP V03-020-1 : Lignes directrices et exigences pour les méthodes basées sur la détection des acides nucléiques
- NF EN ISO 21569 : Méthodes qualitatives basées sur l'utilisation des acides nucléiques
- Norme NF EN ISO 21570 : Méthodes quantitatives basées sur l'utilisation des acides nucléique
- 3 normes liées à l'utilisation de méthode PCR pour la détection des pathogènes dans les aliments :
- Norme NF EN ISO 22174 : Exigences générales et définitions
- Norme NF EN ISO 20837 : Exigences relatives à la préparation des échantillons pour la détection qualitative
- Norme NF EN ISO 20838 : Exigences relatives à l'amplification et à la détection pour les méthodes qualitatives
Le processus de validation
- la solution rêvée... Le laboratoire :
- Utilise une méthode internationale, publiée, validée
- Participe à des essais interlaboratoires, organisés par un organisme indépendant
- Peut montrer que ses propres résultats lors de ces essais interlaboratoires sont corrects
- A mis en place les fondamentaux (système de management de la qualité, formation du personnel, maîtrise de l'environnement, suivi métrologique du matériel, traçabilité des réactifs et des mesures
- Mais si on utilise une méthode complètement développée en interne, ou qu'on utilise une méthode connue, mais adaptéé, ou encore qu'on utilise une méthode connue, mais employée en-dehors de son domaine d'application prévu, alors il faut prouver que la méthode en question est bien apte à l'emploi qu'on prévoir d'en faire.
- Attention : le travail de validation de méthode ne doit pas (ne devrait pas...) être confondu avec le nécessaire travail de développement de méthode, qui constitue l'étape préalable. Néanmoins, il est possible de ré-utiliser, dans le dossier de validation, des résultats obtenus au cours de la phase de mise au point.
- Le processus de validation se déroule alors de la manière suivante :
- On part d'une méthode décrite avec précision, sans ambiguïté,
- On en représente les différentes étapes sous forme de logigramme,
- A chaque étape, on note les matières, réactifs et/ou appareillages critiques (ceux dont la maîtrise est indispensable),
- On explicite clairement les points à valider,
- Pour chacun d'entre eux, on définit une stratégie de validation (manipulations à effectuer, validation par la bibliographie, résultats d'essais interlaboratoires effectués, etc.),
- On définit également une stratégie permettant de valider la manipulation dans son ensemble,
- On prépare les manipulations à réaliser (et en particulier, on dimensionne l'étendue des essais de répétabilité et de reproductibilité),
- On réalise les essais en question,
- Et on analyse les résultats.
- Si tout s'est déroulé conformément aux attentes, la méthode est validée, et on peut écrire le dossier de validation.
Les différents types de méthodes analytiques – et les dossiers de validation associés
- On distingue trois types de méthodes analytiques :
- Méthodes quantitatives (vraies) : elles fournissent un résultat chiffré sur une échelle continue, dont les limites sont connues, en relation directe avec la quantité (ou l'activité) de l'analyte recherché
Leur dossier de validation permettra de statuer sur :- Spécificité
- Justesse
- Sensibilité : limites de détection et de quantification
- Contamination entre échantillons
- Fidélité (répétabilité et reproductibilité)
- Linéarité
- Domaine d'analyse (limites haute et basse)
- Stabilité
- Robustesse
- Valeurs de référence (si possible)
- Interférences
- Corrélation avec la méthode de référence
- Méthodes qualitatives : elles fournissent une information de type présence/absence, ou éventuellement sur une présence supérieure à un témoin. Il existe parfois une part de subjectivité dans la comparaison au témoin positif ou négatif.
Leur dossier de validation permettra de statuer sur :- Spécificité
- Sensibilité
- Contamination entre échantillons
- Stabilité
- Robustesse
- Interférences
- Corrélation avec la méthode de référence
- Méthodes semi-quantitatives : on les assimile à des méthodes quantitatives; elles fournissent un résultat qualitatif extrapolé à partir de la mesure d'une donnée quantifiable (par ex: techniques immuno-enzymatiques avec lecture d'une D.O.). Pour ce type de méthodes, l'incertitude autour du résultat devient une information cruciale lorsqu'il s'agit d'interpréter un résultat ayant une valeur proche de la limite.
Le contenu de leur dossier de validation est intermédiaire entre les deux précédents :- Spécificité
- Sensibilité : limites de détection et de quantification
- Contamination entre échantillons
- Fidélité (répétabilité et reproductibilité)
- Linéarité
- Domaine d'analyse (limites haute et basse)
- Stabilité : si besoin (stabilité des réactifs essentiellement, mais aussi le cas échéant stabilité du signal)
- Robustesse
- Valeurs de référence (si possible)
- Interférences
- Corrélation avec la méthode de référence
- Méthodes quantitatives (vraies) : elles fournissent un résultat chiffré sur une échelle continue, dont les limites sont connues, en relation directe avec la quantité (ou l'activité) de l'analyte recherché
Un petit peu de statistiques
- La détermination des paramètres de répétabilité, de reproductibilité et l'estimation de l'incertitude autour du résultat font intervenir des concepts et des tests statistiques. Il n'est pas question que tous les techniciens d'un laboratoire deviennent des spécialistes en statistiques, mais il est bon qu'au moins une personne au laboratoire possède de solides bases en la matière.
D'ailleurs, lorsqu'il examine un dossier de validation, le COFRAC est sensible au fait qu'un statisticien ait participé à son élaboration. - Sans faire ici un cours de statistiques, je vais rappeler un certain nombre d'incontournables en la matière :
- Hypothèse de normalité : il existe plusieurs méthodes pour tester la normalité d'une distribution :
- La droite de Henry, qui se traçe sur du papier "gausso-arithmétique", et qui s'analyse sans critère (c'est l'œil du statisticien qui permet de décider si les points forment une "bonne" droite...). Elle présente (présentait, à l'époque où les calculettes ou les tableurs n'existaient pas) l'intérêt de donner graphiquement la moyenne et l'écart type).
- Le test du Chi-deux qui, bien qu'il ne soit pas sécifiquement destiné à cela, permet, en comparant la distribution observée à une distribution gaussienne, de même moyenne et de même écart type de valider ou non que les deux sont différentes. Attention, si vous avez des variables continues, définir les valeurs théoriques n'est pas chose aisée.
- Le test de Shapiro et Wilk, spécifiquement destiné à cet usage :
on ordonne les valeurs,
on soustrait la plus petite de la plus grande , on soustrait la deuxième plus petite de la deuxième plus grande, etc.
on multiplie les résultats obtenus par des facteurs de pondération
on fait la somme de ces résultats
on calcule le critère W = carré de la somme précédente/somme des carrés des écarts à la moyenne; le W est comparé à des valeurs seuils dans une table.
- La droite de Henry, qui se traçe sur du papier "gausso-arithmétique", et qui s'analyse sans critère (c'est l'œil du statisticien qui permet de décider si les points forment une "bonne" droite...). Elle présente (présentait, à l'époque où les calculettes ou les tableurs n'existaient pas) l'intérêt de donner graphiquement la moyenne et l'écart type).
- Comparaison de plusieurs séries d'essais : on teste l'homogénéité des variances, grâce au test de Cochran :
Pour chaque laboratoire (ou chaque technicien), on enregistre les résultats des répétitions
On calcule moyenne et écart type
On calcule S = variance = carré de l'écart type
On calcule C = variance la plus élevée / somme des variances
C est comparé à des seuils dans une table
Si les variances ne sont pas homogènes, c'est qu'un au moins des laboratoires (techniciens) ne maîtrise pas la manipulation Ce laboratoire (ce technicien) doit identifier la source de variabilité, avant que l'on puisse estimer la reproductibilité de la manipulation. - Identifier une valeur aberrante : il est toujours difficile d'éliminer a priori une valeur dans une série de données. Et il est parfois difficile d'éliminer une valeur qui semble éloignée des autres, mais qui va dans le sens cherché par l'expérimentateur. Le test de Grubbs est fait pour lever ces doutes :
On ordonne les valeurs
On calcule moyenne et écart type
On calcule l'écart entre la valeur extrême (supérieure puis inférieure) et la moyenne
On calcule G = résultat précédent / écart type de la série
G est comparé à des valeurs seuils dans une table
On peut de même tester si 2 valeurs extrêmes sont aberrantes - Répétabilité : fidélité mesurée avec des résultats d'essai indépendants, obtenus :
- Par la même méthode
- Sur des échantillons identiques
- Dans le même laboratoire
- Avec le même opérateur
- Avec les mêmes équipements
- Reproductibilité : fidélité mesurée avec des résultats d'essai indépendants, obtenus :
- Par la même méthode
- Sur des échantillons identiques
- Dans des laboratoires différents
- Avec des opérateurs différents
- Avec des équipements différents
Quels laboratoires choisir ? Au hasard ! Ne prendre que des labos volontaires reviendrait à sous-estimer l'écart type de reproductibilité.
Combien de laboratoires choisir ? Tout dépend de l'incertitude acceptée sur les résultats d'essais ! Avec 20 labos, si chacun mesure 4 échantillons, on aura, avec un niveau de probabilité de 95 %, un écart type vrai qui ne différera pas de plus de ± 20 % de l'écart type estimé. Avec 20 labos, si chacun ne réalise qu'une mesure, on monte à ± 35 %. Et avec seulement 4 labos, réalisant 2 mesures chacun, on atteint ± 60%
Voir ISO 5725 pour des formules détaillées. - fidélité intermédiaire : Un laboratoire unique n'aura généralement pas la possibilité de réaliser des essais dans les conditions de reproductibilité : un seul lieu, un seul automate... On se placera alors dans les conditions intermédiaires entre répétabilité et reproductibilité, en faisant varier ce qui peut varier : deux techniciens, des lots de réactifs et de consommables différents, des conditions de température différentes, une heure de passage différente (pour valider que l'automate ne dérive pas en cours de série), etc.
- Linéarité : Dans le cas des méthodes quantitatives, c'est l'adéquation à un modèle linéaire. Il existe 3 types principaux de relation entre la concentration C cherchée et la réponse r de l'appareillage :
- Droite passant par l'origine : C = a . r
- Droite avec une ordonnée positive à l'origine C = a . r + b
- Relation exponentielle : ln(C) = d + g . ln(r)
Voir ISO 5725 – 2 pour des exemples
- Incertitude : Elle détermine la précision avec laquelle on peut annoncer un résultat d'analyse. C'est la fidélité Dans le cas ou la mesure est juste (pas d'erreur systématique), et où on a calculé l'écart type de reproductibilité avec un grand nombre de données, distribuées selon un modèle normal.
l'incertitude est souvent donnée comme ± 2 fois l'écart type de reproductibilité.
Si on a des raisons de penser que l'écart type de reproductibilité a été calculé avec des résultats incertains (et donc que des composantes d'erreur systématique ont été sous-estimées), on pourra prendre un facteur ± 3 fois l'écart type de reproductibilité.
Les facteur 2.s ou 3.s découlent de la répartition d'une population normale : 95% se situe dans la zone comprise entre moyenne – 2s et moyenne + 2s (et 99.7% avec moyenne ± 3s). On fait implicitement 2 hypothèses: 1 – le résultat obtenu correspond à la moyenne des résultats que l'on aurait pu calculer si on avait fait de nombreuses déterminations 2 – la dispersion ne varie pas d'une série de manipulations à l'autre.
- Hypothèse de normalité : il existe plusieurs méthodes pour tester la normalité d'une distribution :
La confirmation en routine
- Une fois la méthode validée, on l'utilise en routine. Mais les déviations sont possibles, les erreurs et les accidents aussi. C'est la raison pour laquelle on va ajouter des éléments de validation dans la méthode : on étalonne ou on calibre l'appareil, on ajoute des contrôles ou des témoins positifs et négatifs dans les séries. Ainsi, on s'assure que chaque série est conforme.
- Il faut également continuer à valider que le laboratoire reste compétent, et que les résultats ne dérivent pas. Pour cela, un moyen efficace est la participation à des essais interlaboratoires. Le laboratoire reçoit régulièrement des échantillons à analyser. Il renvoie alors les résultats qui sont traités statistiquement. Tous les participants peuvent ensuite analyser leur résultat en comparaison avec tous les autres.
- Il existe un grand nombre d'organisateurs d'essais interlaboratoires. On parle aussi de "ring test", ou encore de "proficiency testing". Vous trouverez sur le site Internet de l'EPTIS une liste impressionnante de ces organismes, dans tous les domaines et dans de nombreux pays. Impossible à présent d'ignorer que ça existe !
- Le Contrôle National de Qualité organisé en France pour les Laboratoires de Biologie Médicale par l'ANSM, nouvelle version de l'AFSSAPS, n'est pas un système acceptable pour une évaluation externe de la qualité analytique. Tout d'abord parce qu'il n'est pas assez fréquent. Mais surtout parce qu'il ne fait pas bénéficier les biologistes de statistiques sur les résultats.
Ma légitimité dans le domaine
 J'interviens régulièrement depuis plusieurs années sur le sujet, avec des équipes de chercheurs de très haut niveau (Centres Nationaux de Référence et unités INSERM notamment), sur des méthodes devant faire l'objet de publications ou devant être accréditées par le COFRAC.
J'interviens régulièrement depuis plusieurs années sur le sujet, avec des équipes de chercheurs de très haut niveau (Centres Nationaux de Référence et unités INSERM notamment), sur des méthodes devant faire l'objet de publications ou devant être accréditées par le COFRAC.- J'accompagne aussi des laboratoires de biologie médicale qui préparent l'accréditation ISO 15189; il m'arrive assez souvent de réaliser les calculs d'estimation de l'incertitude analytique des méthodes accréditées.
- J'anime des sessions de formation aux Normes ISO 17025 ou 15189 et à la validation de méthodes; et j'accompagne les travaux des équipes.
 Je suis intervenu (en 2012 et 2013) en Côte d'Ivoire, dans le cadre d'un projet financé par le PEPFAR, le Fonds du Président des États-Unis pour la lutte contre le SIDA, et l'American Society for Microbiology, avec l'ONG locale African Medical Laboratory Services. Nous avons ainsi accompagné l'Institut Pasteur de Côte d'Ivoire, le Centre National de Transfusion Sanguine et la structure de lutte contre le VIH, RETROCI, dans leurs projets d'accréditation ISO 15189.
Je suis intervenu (en 2012 et 2013) en Côte d'Ivoire, dans le cadre d'un projet financé par le PEPFAR, le Fonds du Président des États-Unis pour la lutte contre le SIDA, et l'American Society for Microbiology, avec l'ONG locale African Medical Laboratory Services. Nous avons ainsi accompagné l'Institut Pasteur de Côte d'Ivoire, le Centre National de Transfusion Sanguine et la structure de lutte contre le VIH, RETROCI, dans leurs projets d'accréditation ISO 15189.
Par ailleurs, le laboratoire Longchamp, au Plateau, à Abidjan, a été le premier laboratoire privé de Côte d'Ivoire à être accrédité ISO 15189. Je suis heureux d'avoir pu participer à ce succès.- Je travaille régulièrement en partenariat avec quelques Cabinets de Consultants en Management, performance, excellence opérationnelle:
 Le cabinet marseillais Else Consultants, incontournable dès lors que l'on aborde le domaine de la qualité dans les laboratoires de biologie médicale;
Le cabinet marseillais Else Consultants, incontournable dès lors que l'on aborde le domaine de la qualité dans les laboratoires de biologie médicale;
 Le cabinet de conseil en organisation lyonnais Optim Ressources, qui a mené plusieurs Centres Nationaux de Référence en biologie et plusieurs Centres de Ressources Biologiques jusqu'à la certification.
Le cabinet de conseil en organisation lyonnais Optim Ressources, qui a mené plusieurs Centres Nationaux de Référence en biologie et plusieurs Centres de Ressources Biologiques jusqu'à la certification.
 SFC formation consulting, cabinet lyonnais spécialisé dans l'accompagnement de structures du secteur de la santé : hôpitaux, cliniques, pharmacies, laboratoires de biologie médicale.
SFC formation consulting, cabinet lyonnais spécialisé dans l'accompagnement de structures du secteur de la santé : hôpitaux, cliniques, pharmacies, laboratoires de biologie médicale.